メイトペアライブラリーのシーケンスを参照配列にマッピングしてインサート長の分布を調べたい。
このとき、リードの中にアダプターが含まれていて、単純にマッピングすると アダプターを超えて3'側の領域の方がアラインメントスコアが高いprimaryなマッピングとなり、 FRなペアであるように解析されることがあり問題となる。
これを回避するには、あらかじめ、アダプター以降の配列を除去してからマッピングすれば良い。
cutadaptでアダプターを除去する
bwa memを使うこととする。
$ cd $ wget http://cutadapt.googlecode.com/files/cutadapt-1.2.1.tar.gz $ tar zxvf cutadapt-1.2.1.tar.gz
複数のライブラリーを比較する場合、リード数が揃っていないと、ライブラリーの質と、 シーケンシング条件が混ざって分かりにくくなるので、リード数を一定に揃えてから 解析する。
$ cd run_data_dir $ pigz -dc s_1_1_sequence.qf.fastq.gz | head -80000000 | pigz -b 2048 > s_1_1.20M.fastq.gz & $ pigz -dc s_1_2_sequence.qf.fastq.gz | head -80000000 | pigz -b 2048 > s_1_2.20M.fastq.gz
read1とread2の刈り込みは一方をバックグラウンドで同時並列におこなうことが出来る。
CPUとdisk IOの能力のバランスでgzipした方が良いかどうかは異なる。ここでは、 CPUの余裕の方が多い状況を仮定して、入出力はpigzによってgzip形式に圧縮する例を示しているが、 IOに余裕がある場合は、圧縮しない方が速い。
cutadaptのオプションについては README fileに説明されている。
ここでは、-a オプションを用いて検出されたアダプター配列の3'側をとりのぞく。
$ ~/cutadapt-1.2.1/bin/cutadapt -a CTGTCTCTTATACACATCT -a AGATGTGTATAAGAGACAG -a GATCGGAAGAGCACACGTCTGAACTCCAGTCAC s_1_1.20M.fastq.gz | pigz -b 2048 > s_1_1.20M.trim.fastq.gz & $ ~/cutadapt-1.2.1/bin/cutadapt -a CTGTCTCTTATACACATCT -a AGATGTGTATAAGAGACAG -a GATCGGAAGAGCACACGTCTGAACTCCAGTCAC s_1_2.20M.fastq.gz | pigz -b 2048 > s_1_2.20M.trim.fastq.gz
$ bwa mem -t 8 ~/chara0.clean.contigs.fasta s_1_1.20M.trim.fastq.gz s_1_2.20M.trim.fastq.gz | samtools view -S -b - > mp1.20M.trim.bam
bwaはSAMを標準出力に出力するのでそのままパイプでsamtools viewに渡してbam fileにする。
$ samtools view -H mp1.20M.trim.bam > samhead
$ samtools view mp1.20M.trim.bam | ruby -e 'ARGF.each_line{|line| s=line.split("\t")[9]; puts line if s!=nil && s.size>10}'|cat samhead - | samtools view -bS - > mp1.20Mlong.bam
$ java -jar ~/lcl/lib/picard-tools-1.92/SortSam.jar I=mp1.20Mlong.bam O=mp1.20M.trimaligned.bam SORT_ORDER=coordinate
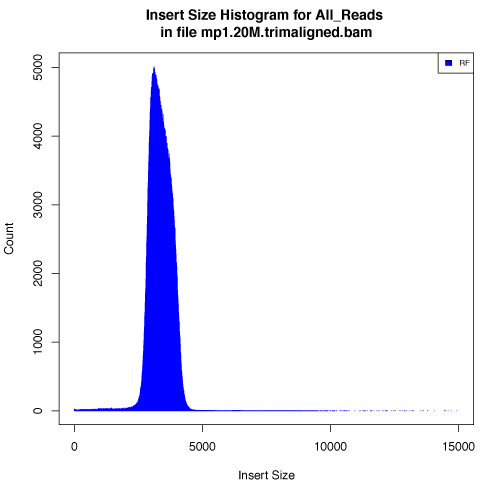
$ java -jar ~/lcl/lib/picard-tools-1.92/CollectInsertSizeMetrics.jar I=mp1.20M.trimaligned.bam O=mp1.5M.trim.metrics H=mp1.20M.trim.pdf HISTOGRAM_WIDTH=15000これによって次のようなヒストグラムが得られる。
上のヒストグラムは、1つのライブラリーのインサート長を表現するには十分であるが、 複数のライブラリーのインサート長を比較するには、塗りつぶされていることが災いして 分かりにくいものとなる。
そこで、インサート長と頻度の標からRを用いてプロットすることとする。
次の様にしてインサート長と頻度の対応表取り出すことが出来る。
% sed -n -e '/^insert_size/,$p' > mp1.20M.insertsizefreq
この後Rのコンソールから読み取って、自由にグラフを重ねることができる。
% R
...
> mp1 <-read.table("mp1.20M.insertsizefreq", head=T)
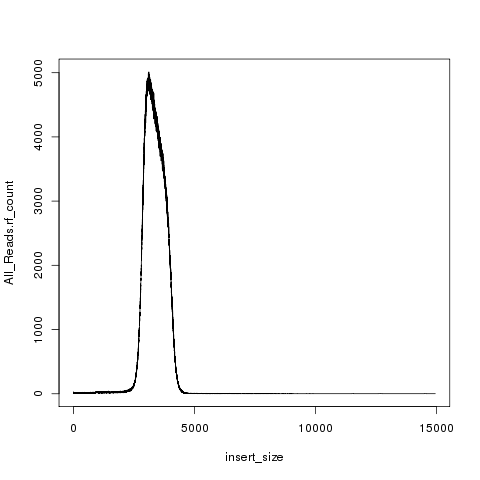
> plot(mp1, type="l")
これによって次のようなfrequency polygonが得られる。
図の基本構造に満足したら、PNGやPDF形式のファイルに書き出す。例えばPNGに書き出すには
> png("mp1.20M.insertsize.png")
> plot(mp1, type="l")
> dev.off()
PDFに書き出すには
> pdf("mp1.20M.insertsize.pdf")
> plot(mp1, type="l")
> dev.off()
とする。
先の自動で作られたヒストグラムの図と異なり、frequency polygonは塗られていないので重ねあわせ比較が容易である。 col=2やlty=2というようなパラメータを追加することで異なる色、線種(破線,点線等)の設定が可能である。
mp1<- read.table("mp1.5M.trim.metrics.tab", head=T)
plot(mp1,type="n", col=3)