Genome size can be calculated by counting k-mer frequency of the read data. The k should be sufficiently large that most of the genome can be distinguished. For most eukaryotic genomes at least 17 are usually used and calculation upto 31 is easiliy doable with Jellyfish.
Here we assume the following are available.
In this example we have 5 pair of fastq files in three different subdirectories. The file to process can be specified with "*/*.qf.fastq" and veriied with ls.
$ ls */*.qf.fastq run1/s_1_1_sequence.qf.fastq run2/s_2_2_sequence.qf.fastq run1/s_1_2_sequence.qf.fastq run3/s_1_1_sequence.qf.fastq run2/s_1_1_sequence.qf.fastq run3/s_1_2_sequence.qf.fastq run2/s_1_2_sequence.qf.fastq run3/s_2_1_sequence.qf.fastq run2/s_2_1_sequence.qf.fastq run3/s_2_2_sequence.qf.fastq
Next, we issue the jellyfish count command
jellyfish count -t 8 -C -m 25 -s 5G -o spec1_25mer --min-quality=20 --quality-start=33 */*.qf.fastq
First confirm that you got the output file
$ ls spec1_25mer* spec1_25mer_0
now that there is a single file spec1_25mer_0
$ jellyfish histo -o spec1_25mer.histo spec1_25mer_0
Confirm that you got the output
$ ls spec1_25mer* spec1_25mer_0 spec1_25mer.histo
Examine the numbers by your eyes
$ head -25 spec1_25mer.histo 1 461938583 2 95606044 3 19280477 4 13836754 5 11018480 6 9555090 7 8557935 8 7863244 9 7319505 10 6920880 11 6589723 12 6321923 13 6148638 14 6036120 15 5972264 16 5962234 17 5987696 18 6051171 19 6154429 20 6297373 21 6485135 22 6700579 23 6932570 24 7217627 25 7533211
$ R
...
> spec1_25 <- read.table("spec1_25mer.histo")
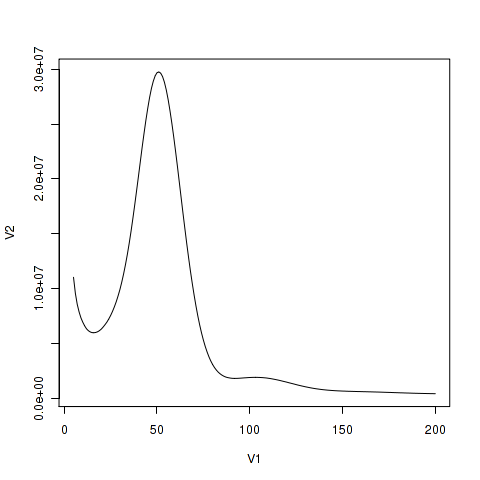
> plot(spec1_25[5:200,],type="l")This will plot from 5 th line in the histo file. In general, the very low frequency k-mer are extremely high number that would make the scale for the y-axis too large. For x values, selection of the higher bound should be determined in accordance with read depth. The data contains upto 10000. However if you plot them, it would be too large. Thus some good number should be chosen.
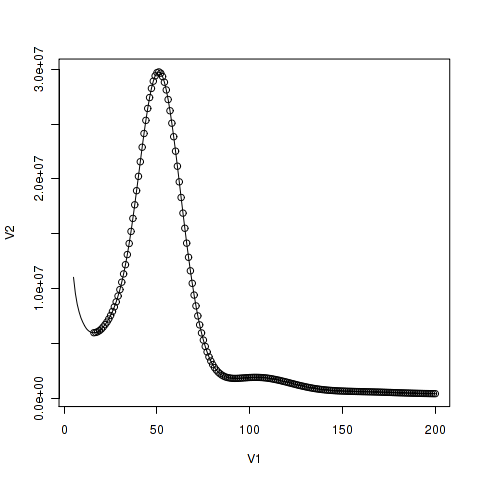
First, you should determine the border between the peak corresponding to single copy region and the initial peak corresponding to sequence errors. You might plot points on the line so that the region would be obvious.
> points(spec1_25[16:200,])
Now, we would calculate the total number of k-mer in the distribution
> sum(as.numeric(spec1_25[16:10000,1]*spec1_25[16:10000,2])) [1] 120107242393
In this case there are about 120 G k-mers in the histogram.
Next, we want to know the peak position. From the graph, we can see its close to 50. Thus we examine the number close to 50 and find the maximum value
spec1_25[40:60,] V1 V2 40 40 20225459 41 41 21557020 42 42 22881520 43 43 24132924 44 44 25331980 45 45 26419451 46 46 27411481 47 47 28246677 48 48 28903809 49 49 29388497 50 50 29680473 51 51 29751275 52 52 29636175 53 53 29324357 54 54 28809699 55 55 28107532 56 56 27232397 57 57 26208735 58 58 25075370 59 59 23840233 60 60 22522009
In this example, the peak is at 51. Then, the genome size can be estimated as:
> sum(as.numeric(spec1_25[16:10000,1]*spec1_25[16:10000,2]))/51 [1] 2355043968
This reads as 2.36 Gb.
If we regard single copy region as frequency between 16 and 80, the size of single copy region can be roughly calculated as
> sum(as.numeric(spec1_25[16:80,1]*spec1_25[16:80,2]))/51 [1] 954755030
It is 0.95 Gb or 40.5% of the genome. The proportion could be calculated as:
> (sum(as.numeric(spec1_25[16:80,1]*spec1_25[16:80,2]))/sum(as.numeric(spec1_25[16:10000,1]*spec1_25[16:10000,2])) [1] 0.4054086
For an inbred line, two set of genomes have mostly identical sequence and the major peak would be diploid (2C) peak. On the otherhand, for highly heterozygous sample, there would be heterozygous peak with 1C depth.
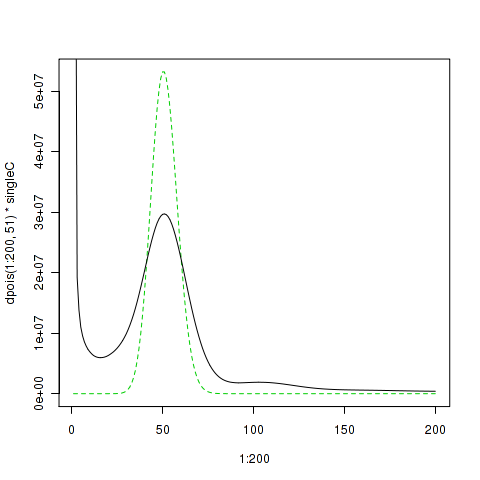
Now that we have some nice curve, we could compare it to ideal curve as poisson distribution scaled to the estimated single copy region size
> singleC <- sum(as.numeric(spec1_25[16:80,1]*spec1_25[16:80,2]))/51 > plot(1:200,dpois(1:200, 51)*singleC, type = "l", col=3, lty=2) > lines(spec1_25[1:200,],type="l")