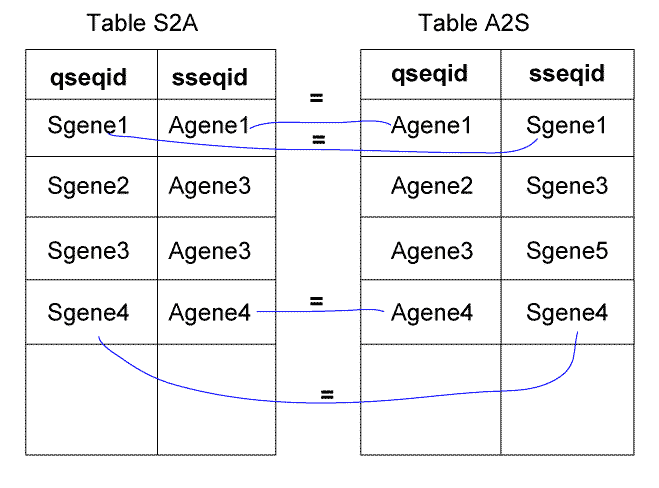
図: S2AとA2Sのtableにおいて、双方向ベストヒットにあたる関係について、一致する要素を青の線で示した。
第5回インフォマティクス情報交換会ではReciprocal Blast Best Hitを抽出する実習を行った。ただし、Reciprocal Best Hitが良い方法であるからではなく簡単だからである点には留意願いたい。 Reciprocal Best Hitなので、調べたい種Sの配列データと良くアノテーションされている参照種Aの双方向のBLAST検索を行い、相互に最上位に来る組を選択すればよい。
実習には、Windowsがインストールされたノートパソコンを用いるという設備環境に対応しつつ、Linuxでの操作を行うため、Scientific LinuxのLiveUSBシステム を用意して行った。LiveUSBの作り方については別ページで解説するが、RDBMSのPostgreSQLを起動してる点、サンプルデータを含めてある点、ライブユーザのホームディレクトリにrubyとbiorubyをインストールしてある点が通常版との違いである。
LiveUSBで起動するとデスクトップ画面に達するが、ここでターミナルを用いる。
NCBIのBLASTの実行ファイルはLiveUSBのトップディレクトリにncbiというディレクトリに入れてあるアーカイブに含まれているので、 これを展開し、中の実行ファイルを ~/binにコピーします。これにより、blastx などのコマンドを使えるようになります。
$ ls /mnt/live $ ls /mnt/live/ncbi $ tar zxvf /mnt/live/ncbi/ncbi-blast-2.2.5+-x64… $ mv ncbi-blast+-2.2.25/bin/* bin
自動補完: 長いファイル名を全部タイプすると間違いがち 他と区別できる程度にタイプしたら「Tab」を押すと自動的に 補完されます。途中から変わる似た名前のファイルがある時は 一致している範囲で補完されます。(bash, tcsh, zshなどの機能)
$ mv ncbi-blast-2.2.25+/bin . $ ls bin $ blastx -help $ makeblastdb -help
>entry_id1 definition of the gene product etc. ACGAAATGAGAGATAT AGAGAGATGCCATAGATAG ATGCAG >entry_id2 definition of the gene product 2 etc. ACGAAATGAGAGATAT AGAGAGATGCCATAGATAG ATGCAG …
ls ls /mnt/live/data
ページャのlessというコマンドを使うとファイルの中身を見る事ができます。基本的な使い方は
less filename
のように内容を見たいファイル名をコマンドに続けて指定します。より具体的には、例えば
less /mnt/live/data/Spur5.fasta
のようにします。
lessが開発される前には、moreという1ページずつ進む事だけができるページャがありました。 lessは進む事しかできないmoreの反対で戻る事ができるページャです。
$ cp /mnt/live/data/Spur5.fasta . $ cp /mnt/live/data/TAIR10_pep_20110103_representative_gene_model . $ makeblastdb -in TAIR10_pep_20110103_representative_gene_model -title TAIR10peprep -taxid 3702 $ makeblastdb -in Spur5.fasta -title SpurRNAseq -taxid 45176 -dbtype nucl
blastx -query Spur5.fasta -db TAIR10_pep_20110103_representative_gene_model -out S2A.asn -outfmt 11
で数時間待てば結果が得られる。しかし、実習時間は限られているので個別のエントリーを取り出していろいろな検索を試すこととする。 以下のコマンドをタイプすると、1つ1つのエントリーが単一のファイルとして1つのディレクトリーに100個ずつ必要な個数のディレクトリーに分割して生成される
ruby /mnt/live/ruby/flatsplit.rb Spur5.fasta 1
その後
blastx -query Spur5.fasta.1/Spur5.fasta.33 -db TAIR10_pep_20110103_representative_gene_model
とすると33 番目のエントリーをqueryにしたBLAST検索が行われ、標準的な形式の出力がターミナルに表示されます。 ただ、この出力は行数が多いので、出力の大部分は画面の上の方に流れさってしまいます。 この出力は標準出力というものにでているので、リダイレクトする事ができます。例えば、
blastx -query Spur5.fasta.1/Spur5.fasta.33 -db TAIR10_pep_20110103_representative_gene_model | less
とするとページャのlessの入力とする事ができ、1画面分単位で見ていく事ができます。
ASN.1形式には基本的な情報がいろいろ格納されており、blast_formatterを使うことによって異なった形式 で表示する事ができます。例えば
blast_formatter -archive S2A.asn -max_target_seqs num_sequences 3 -outfmt 6
などとすることにより、上位3位以内のhitのみを表形式で取り出すという様な事が可能です。
表形式のデータがあるときに最上位のみをとるには例えば、
lastq=''
ARGF.each_line do |line|
ary=line.split
qseqid=ary[0]
if qseqid != lastq
puts line
lastq=qseqid
end
end
$ruby tabrank.rb /mnt/live/data/A2Spur5.tab > A2S.top $ruby tabrank.rb /mnt/live/data/Spur5toA.tab > S2A.top
$ gedit tabrank.rb
BLASTの結果を元に、双方向のbest hitを抽出するには、リレーショナルデータベースを利用するのが有効である。
RDBMSを利用することにより、条件を記述するだけで、実際の選択方法を書かなくても双方向best hitなどの条件に適合する組み合わせを そこそこの時間で抽出する事ができる。
リレーショナルデータベースはtableの集合に対してSQL (Structured Query Language)で問い合わせを行うという使い方をするのが一般的である。
このようにして使うデータベースには
などをはじめ、様々なものがあるが、ここではPostgreSQLを例に用いる。
まず、作るべき表の構造を定義する必要があるが、入力に用いるblastの出力にどのような要素が並んでいるかに会わせた定義をする。 BLASTの出力表の要素は、
blastx -help
の出力を調べると、
'qseqid sseqid pident length mismatch gapopen qstart qend sstart send evalue bitscore'
の順であるとわかるので、これに、それぞれのデータの種類に合わせた指定を加えて、
CREATE TABLE A2S (qseqid text, sseqid text, d float8, length int4, m int4, g int4, qs int4, qend int4, sstart int4, send int4, evalue float8, bitscore float8); CREATE TABLE S2A (qseqid text, sseqid text, d float8, length int4, m int4, g int4, qs int4, qend int4, sstart int4, send int4, evalue float8, bitscore float8);
のような定義を行えば良いとわかる。このように長いコマンドを正しくタイプするのは難しいので、一旦エディターで上記の内容が含まれるファイルcreatetable.psqlを作成し
$ psql < createtable.psql
PostgreSQL databaseとやり取りするためのフロントエンドをinteractiveに利用するように起動する。
$ psql
先のコマンドにリダイレクトしないだけであるが、これにより、キーボードからの入力を受け付けるようになり、 「databasename=>」というプロンプトがあらわれる。
これに対し
\copy A2S from 'A2S.top'
と入力することにより、'A2S.top'というファイルにあった内容がtable A2Sに格納される。同様に、
\copy S2A from 'S2A.top'
により、'S2A.top'というファイルにあった内容がtable S2Aに格納される。
AgeneとSgeneが互いに双方向でbest hitであるというのがreciprocal best hitの関係であるが、 これはAgeneをqueryにした時にのhitにSgeneがあり、Sgeneをqueryにしたbest hitにAgeneがあるということである(図)から、 この条件を書き下すと、
S2A.qseqid = A2S.sseqid and S2A.sseqid = A2S.qseqid
というように表せる。
図: S2AとA2Sのtableにおいて、双方向ベストヒットにあたる関係について、一致する要素を青の線で示した。
SQL文全体としては
SELECT A2S.qseqid as a2sq, S2A.qseqid as s2aq from A2S, S2A where S2A.qseqid = A2S.sseqid and S2A.sseqid = A2S.qseqid;
となる。これをpsqlのプロンプトに対して入力すると、実際に組み合わせが出力される。 しかしながら、その結果は表示されるだけである。これを、記録として取得するには、結果を新たなtableに格納すれば良い。すなわち
SELECT A2S.qseqid as a2sq, S2A.qseqid as s2aq into reciprocalbest from A2S, S2A where S2A.qseqid = A2S.sseqid and S2A.sseqid = A2S.qseqid;
のようにinto reciprocalbestを加えることにより、reciprocalbestというtableに結果が全て格納される。このテーブルに対して検索を行う事ができる。 たとえば、
Select count(*) from reciprocalbest;
と問い合わせる事で、この表には何行有るかがわかる。この場合は11622行あるという結果が得られた。