JBrowseのインストールに関する覚え書き
西山智明 tomoakin@staff.kanazawa-u.ac.jp
2014年6月2日
要点:
- Augustusからのgff3をimportする時に--type=transcriptをつける。
- .bam, .baiにcontent-type: application/octet-streamを設定する。
- イントロンをまたぐリードの色がおかしいのでpull requestを提出した。
JBrowse-1.11.4.zipをダウンロードして展開
http://jbrowse.org/ のLatest Releaseをダウンロードして展開する。リンクURLをコピーしてwgetすると。
$ wget http://jbrowse.org/wordpress/wp-content/plugins/download-monitor/download.php?id=95 --2014-06-02 22:00:57-- http://jbrowse.org/wordpress/wp-content/plugins/download-monitor/download.php?id=95 Resolving jbrowse.org... 184.72.219.192 Connecting to jbrowse.org|184.72.219.192|:80... connected. HTTP request sent, awaiting response... 302 Found Location: http://jbrowse.org/releases/JBrowse-1.11.4.zip [following] --2014-06-02 22:00:58-- http://jbrowse.org/releases/JBrowse-1.11.4.zip Connecting to jbrowse.org|184.72.219.192|:80... connected. HTTP request sent, awaiting response... 200 OK Length: 5520117 (5.3M) [application/zip] Saving to: "download.php?id=95" 100%[=========================================================================================>] 5,520,117 1.14M/s in 6.4s 2014-06-02 22:01:05 (848 KB/s) - "download.php?id=95" saved [5520117/5520117] $ mv download.php\?id\=95 JBrowse-1.11.4.zip $ unzip JBrowse-1.11.4.zip Archive: JBrowse-1.11.4.zip creating: JBrowse-1.11.4/ creating: JBrowse-1.11.4/bin/ inflating: JBrowse-1.11.4/bin/add-bam-track.pl inflating: JBrowse-1.11.4/bin/add-bw-track.pl ... inflating: JBrowse-1.11.4/tristate.css finishing deferred symbolic links: JBrowse-1.11.4/sample_data/json/modencode/tracks/volvox_microarray.wig -> ../../volvox/tracks/volvox_microarray.wig JBrowse-1.11.4/sample_data/raw/Dmel.json -> ../../docs/examples/config/Dmel.json JBrowse-1.11.4/sample_data/raw/volvox -> ../../docs/tutorial/data_files JBrowse-1.11.4/sample_data/raw/volvox.json -> ../../docs/tutorial/conf_files/volvox.json $ mv JBrowse-1.11.4 jbrowse
という具合になった。ここから、wget http://jbrowse.org/releases/JBrowse-1.11.4.zipで取得できることがわかるが、再度とっても同じ事なので改名するだけで十分だろう。展開したらJBrowse-1.11.4というdirectoryができるが、運用用にバージョン無しのjbrowseに改名した。
setup.shを実行
$ sh ./setup.sh Installing Perl prerequisites ... done. Formatting Volvox example data ... done. To see the volvox example data, browse to http://your.jbrowse.root/index.html?data=sample_data/json/volvox. Formatting Yeast example data ... done. To see the yeast example data, browse to http://your.jbrowse.root/index.html?data=sample_data/json/yeast. Building and installing legacy wiggle format support (superseded by BigWig tracks) ... done. Building and installing legacy bam-to-json.pl support (superseded by direct BAM tracks) ... done.
なんか、perlのモジュールが足りないというメッセージがでたらcpanからインストールする。
apache2の設定
/etc/httpd/conf.d/Jin03_jbrowse.conf に
Alias "/Jin03/jbrowse" "/ssd4/jbrowse"
の一行を加えて、httpdを再起動する。これで、web browserからのアクセスが可能になる。
http://ip.address.example.com/Jin03/jbrowse/docs/tutorial/ からtutorialが読めるようになる。
さらに「 Volvox test data」のリンクを辿って"volvox"データをみて確認することができる。最初に開いた時点ではほぼ白紙であるが、左のGFF3などのチェックボックスをチェックすると遺伝子モデルが現れる。なお、このvolvoxと言う名前はGBrowse以来であるが、シークエンスされたVolvoxとは全く関係がないダミーデータである。
参照配列の読み込み
$ bin/prepare-refseqs.pl --fasta ../Jin03/Jin03.fa
annotationの読み込み
$ bin/flatfile-to-json.pl --gff ../Jin03/aug3.gff3 --type transcript --trackLabel aug3tr --trackType CanvasFeatures $ bin/flatfile-to-json.pl --gff ../Jin03/aug3.gff3 --trackLabel aug3 --trackType CanvasFeatures
gff3のファイルを読み込む。 注意点は、--type=transcript をつけないとintronを細い線でCDSを太くするのが実行されず、遺伝子全体が1つの四角で表示される。
BAM ファイルを表示できるようにする
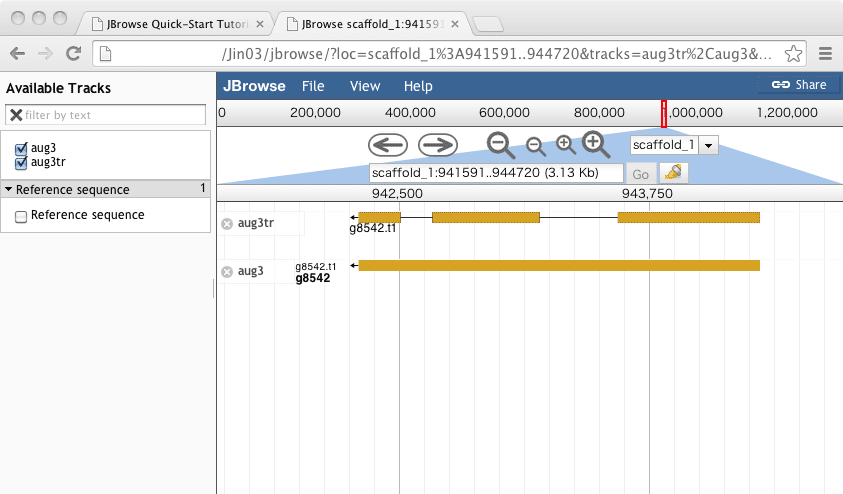
次にRNA-seqのデータを遺伝子モデルとセットで見られるようにしたい。マッピング済みデータはあるのでbam fileをもってきてみれば良いというところである。
tracks.confの編集
# make a section for the new track called tracks.(unique name) [ tracks.RNA-seq-bam-track1 ] #Sample_idx2_thm # settings for what data is shown in the track storeClass = JBrowse/Store/SeqFeature/BAM urlTemplate = Sample_idx2_thm/accepted_hits.bam baiUrlTemplate = Sample_idx2_thm/accepted_hits.bam.bai # settings for how the track looks category = NGS #< category for this track type = JBrowse/View/Track/Alignments2 key = BAM idx2 maxHeight=450
の様なデータをサンプル毎に繰り返し用意し。bam fileとindexをdata/の下に Sample_idx2_thm/accepted_hits.bam, Sample_idx2_thm/accepted_hits.bam.baiとして置く。これだけで良いはずである。
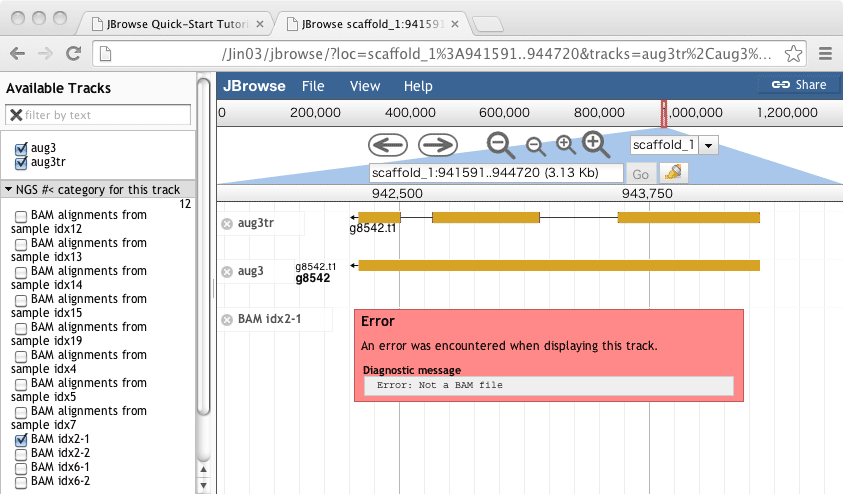
やってみると、何故かBAMが壊れているというメッセージがでる。Volvoxの例もBAMを見ようとすると、Error Not a BAM fileというエラーメッセージが現れる???
さらにアクセスログを/var/log/httpd/access_log にて確認すると
XXX.XXX.XX.XX - - [02/Jun/2014:22:40:18 +0900] "GET /Jin03/jbrowse/data/Sample_idx2_thm/accepted_hits.bam HTTP/1.1" 206 65536 "http://XXX.XXX.XX.XX/Jin03/jbrowse/?loc=scaffold_1%3A941591..944720&tracks=aug3tr%2Caug3%2CRNA-seq-bam-track1f&highlight=" "Mozilla/5.0 (Macintosh; Intel Mac OS X 10_6_8) AppleWebKit/537.36 (KHTML, like Gecko) Chrome/35.0.1916.114 Safari/537.36"
というのが見つかる。
では、ここで転送しようとしているBAM ファイルをhttpで取得できるか試すと問題なく取得できる。。。。
.bam, .baiへのmime-type設定
curlで確認してみるとどうも、bamファイルのcontent-transfer-encodingやbaiのcontent-typeがあやしい。
$ curl -I http://XXX.XXX.XX.XX/Jin03/jbrowse/data/Sample_idx2_thm/accepted_hits.bam HTTP/1.1 200 OK Date: Mon, 02 Jun 2014 13:50:19 GMT Server: Apache/2.2.15 (CentOS) Last-Modified: Mon, 12 May 2014 02:53:30 GMT ETag: "1cc0200-43917f7a-4f92b0d5dbe80" Accept-Ranges: bytes Content-Length: 1133608826 Connection: close Content-Type: application/octet-stream Content-Encoding: x-gzip $ curl -I http://133.28.110.70/Jin03/jbrowse/data/Sample_idx2_thm/accepted_hits.bam.bai HTTP/1.1 200 OK Date: Mon, 02 Jun 2014 13:50:54 GMT Server: Apache/2.2.15 (CentOS) Last-Modified: Sun, 01 Jun 2014 08:23:03 GMT ETag: "1cc0205-9f5a8-4fac1fcc692af" Accept-Ranges: bytes Content-Length: 652712 Connection: close Content-Type: text/plain; charset=UTF-8
そこで、apache2の設定を調整して、mime-typeをapplication/octet-streamになるように設定する。すなわち、
AddType application/octet-stream .bai AddType application/octet-stream .bam
を/etc/httpd/conf.d/Jin03_jbrowse.conf に加えて、httpdを再起動する。リロードしても、改善しないが、くじけずに、さらに、browserのキャッシュをクリアしてからリロードすると、アラインメントが見えるようになった。
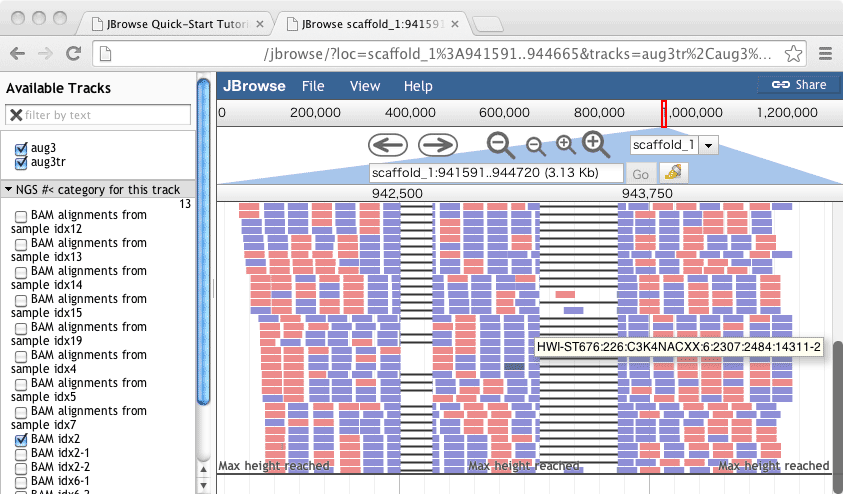
イントロンをまたぐRNA-seqのリードのstrand評価がおかしい点への対処
さて、めでたく表示できるようになった。せっかくだから、read 1, read2にデータをわけてstranded RNA-seqが正しく行われていることを見ようかと思い、次に示すseparate_reads.shを用いて分離したbamファイルを用意し、tracks.confを編集して対応するtrack情報を作った。
$ cat separate_reads.sh #!/bin/bash samtools view -H accepted_hits.bam > head cp head accepted_hits-1.sam cp head accepted_hits-2.sam samtools view accepted_hits.bam |grep -- '-1 ' >> accepted_hits-1.sam & samtools view accepted_hits.bam |grep -- '-2 ' >> accepted_hits-2.sam & wait samtools view -bS accepted_hits-1.sam > accepted_hits-1.bam && samtools index accepted_hits-1.bam & samtools view -bS accepted_hits-2.sam > accepted_hits-2.bam && samtools index accepted_hits-2.bam wait
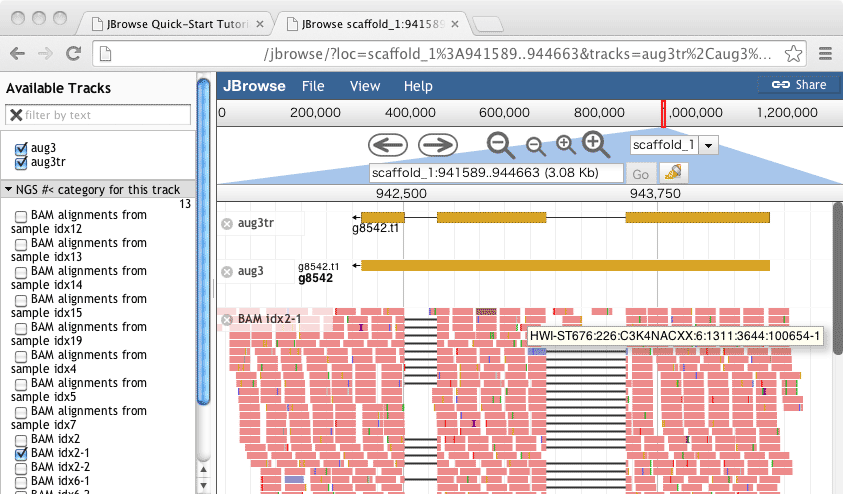
やってブラウザでみると、どうも、イントロンをまたぐリードだけ色がおかしい。
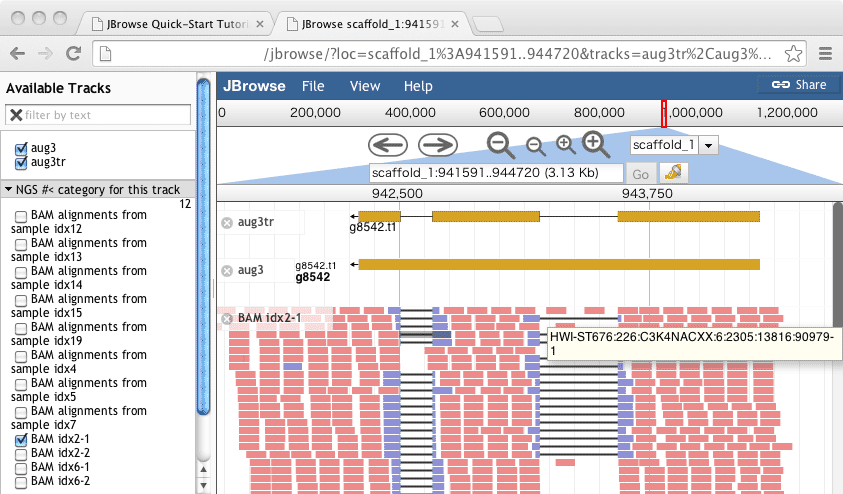
詳しい情報をみて、元のsam recordと見比べてもstrandの認識が逆になっているようである。
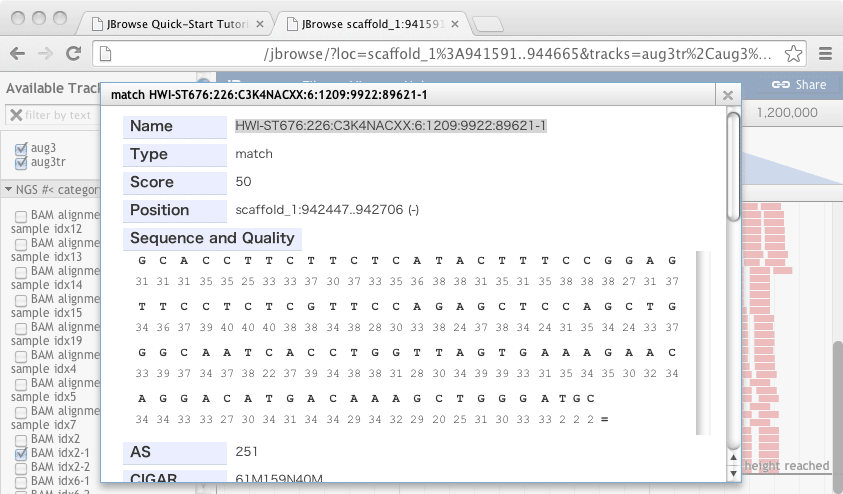
$ samtools view accepted_hits-1.bam | grep HWI-ST676:226:C3K4NACXX:6:1209:9922:89621-1 HWI-ST676:226:C3K4NACXX:6:1209:9922:89621-1 0 scaffold_1 942447 50 61M159N40M * 0 0 GCACCTTCTTCTCATACTTTCCGGAGTTCCTCTCGTTCCAGAGCTCCAGCTGGGCAATCACCTGGTTAGTGAAAGAACAGGACATGACAAAGCTGGGATGC @@@DD:BBF?FBDEGG@D@DGGG<@FCEFHIIIGGCG=?BG9FGC9@DC9BFBHFCFG7FHCGG@=?CHHB@DCD?ACCCBB<?C@CC>CA>5:@?BB### AS:i:-5 XM:i:1 XO:i:0 XG:i:0 MD:Z:0A100 NM:i:1 XS:A:- NH:i:1
ブラウザ上で、Positioが(-)になっているが元のSAMレコードを見てもFLAGは0であり、reverse complementにはなっていない。
strandに関わりそうなコードを探すと、src/JBrowse/Store/SeqFeature/BAM/LazyFeature.js というファイルに
strand: function() {
var xs = this._get('xs');
return xs ? ( xs == '-' ? -1 : 1 ) :
this._get('seq_reverse_complemented') ? -1 : 1;
},というコードがあってこれで決めているらしい。さらに前後xs, seq_reverse_complementedを調べると、XS fieldが設定されている時はXSに従い、ないときはFLAGに従うということである。では、XSというのがどういうものであるかというと、intronのsplice siteの情報にもとづいて、そのリードがtranscriptに対してsenseであるかanti senseであるかを推定したflagであるらしい。そしてFLAGの参照配列に対して+鎖か-鎖かというのとは意味合いが異なる事がわかりごた混ぜにするのは良くないと考えられる。この点をgithubのpull requestとして提出した。https://github.com/GMOD/jbrowse/pull/485
実際に送られるのは、スペースを圧縮されたsrc/JBrowse/Store/SeqFeature/BAM.jsなのでその当該部を編集することでそのまま修正できる。圧縮されているが、「var a=this._get("xs");return a?"-"==a?-1:1:」を「return 」で置き換えればよい。こうしてほぼRNA-seqのデータがstrandが揃っているのが見えるようになった。