本稿では、ゲノムにマッピングしたデータにもとづいてインサート長のグラフを作成する例を紹介する
なるべく標準的な既存のツールとしてpicardを使うこととする。
picardはjava環境で動作するソフトウエアである。java環境はMac OS附属のもので問題なく動く。 毎回ではないが、最初にpicardを入手する。 picardのホームページはhttp://picard.sourceforge.net/である。 適切な(近くの)ミラーサイトから 最新版をダウンロードし保存する。
「ダウンロード」フォルダに保存したら、ターミナルからはDownloadsに見えるので
% unzip Downloads/picard-tools-1.92.zip
のようにして展開する。
概略としては以下のようになる
ここでは、PhiX1Mpebwa.samというファイルを例に示す
% samtools view -bS PhiX1Mpebwa.sam > PhiX1Mpebwa.bam [samopen] SAM header is present: 1 sequences.
これにより、PhiX1Mpebwa.samの内容を持つbam形式のファイル PhiX1Mpebwa.bamが出来る
前節で作成したファイルをさらにソートする
% samtools sort PhiX1Mpebwa.bam PhiX1Mpebwa-sorted
これによってPhiX1Mpebwa-sorted.bamというファイルができる
% java -jar ~/picard-tools-1.92/CollectInsertSizeMetrics.jar INPUT=PhiX1Mpebwa-sorted.bam O=PhiX1M.sam.insertsizemetrics H=PhiX1M.sam.hist.pdf
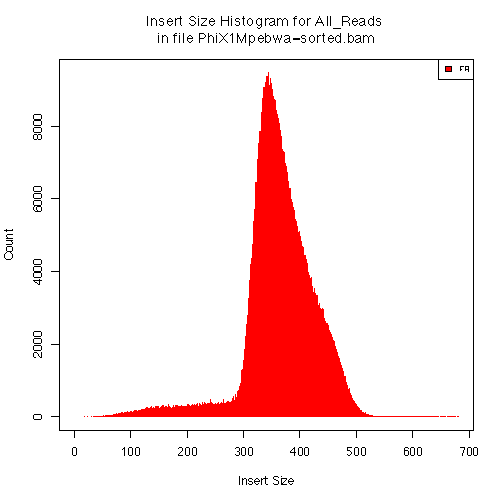
これにより、PhiX1M.sam.insertsizemetrics というテキストファイルとPhiX1M.sam.hist.pdf というpdfファイルができる。PhiX1M.sam.hist.pdfは下に示すようなヒストグラムになっている。
次の様にしてインサート長と頻度の対応表取り出すことが出来る。
% sed -n -e '/^insert_size/,$p' PhiX1M.sam.insertsizemetrics > PhiX1M.sam.insertsizefreq
この後Rのコンソールから読み取って、自由にグラフを重ねることができる。
% R
...
> PhiXIns <-read.table("PhiX1M.sam.insertsizefreq", head=T)
> plot(PhiXIns, type="l")
これによって次のような図が得られる。
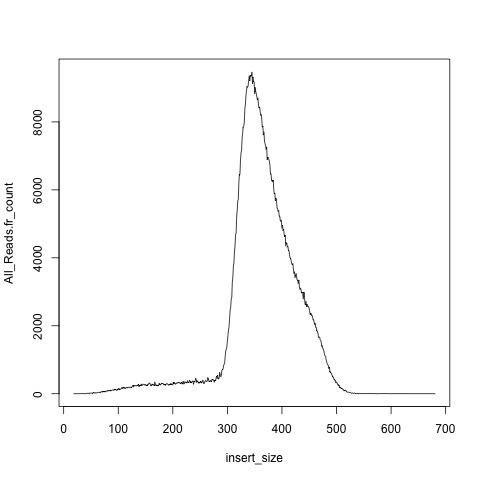
先の自動で作られた図と異なり、塗られていないので重ねあわせ比較が容易である。
col=2やlty=2というようなパラメータを追加することで異なる色、線種(破線,点線等)の設定が可能である。